ATAC-Seq(Assay for Transposase-Accessible Chromatin with high throughput sequencing) 是2013年由斯坦福大学的研究人员开发的用于研究染色质可及性(通常也理解为染色质的开放性)的方法,原理是通过转座酶Tn5容易结合在开放染色质的特性,然后对Tn5酶捕获到的DNA序列进行测序。ATAC-Seq由于所需细胞量少,实验简单,可以在全基因组范围内检测染色质的开放状态,已经成为研究染色质开放性的首选技术方法。 目前,该技术被用于测定特定时空下基因组中所有处于开放状态的序列,分析调控元件,揭示转录因子结合位点以及核小体位置,从而为研究精细化基因表达的调控、DNA印记等提供有效的方法。


胰岛素分泌受损是2型糖尿病(T2D)的标志,而染色质结构的改变可能导致这种疾病。因此,本案例使用ATAC测序剖析T2D和非糖尿病供体胰岛的染色质开放区域,以研究T2D对人类胰岛开放染色质的影响。
瑞典乌普萨拉大学北欧胰岛移植网络的9例非糖尿病供体(对照组)和6名被诊断为T2D供体(实验组)
分离、培养胰岛细胞;取约30个新鲜胰岛冷冻(5个供体)或立即用于ATAC测序(5个供体);测序数据整理分析;胰岛的RNA-seq;ATAC-seq数据与公共ChIP-seq数据集联合分析。
1. 绘制人类胰岛开放染色质图谱
非糖尿病和T2D组中分别鉴定出57,105和53,284个peaks,一些序列被富集到与胰岛发育分化有关的基因上,例如PDX-1和FOXA2(图1)。其中,大部分peaks的位置接近转录起始位点(TSS)(图2a),在糖尿病和非糖尿病供体的胰岛中,染色质开放区域的基因组分布相似(图2b, c)。
2. 增强子元件和组蛋白修饰注释分析
与活性染色质相关的组蛋白标记(H3K4me1、H3K4me3和H3K27ac)在实验组胰岛peak中富集,但只有一小部分异染色质相关的组蛋白标记(H3K27me3和H3K9me3)与peak重叠(图3),且T2D供体胰岛中的组蛋白修饰和peak之间的重叠与之类似。此外,T2D相关的差异性甲基化区域(DMRs)位于人类胰岛的7412个开放染色质区域,包括PDX1和SLC20A2标注的区域。
3. 转录因子和转录本注释分析
通过ATAC-seq开放染色质区域和胰岛特异性转录因子(TF)相关基因的比对分析,发现研究所涉及的TF相关基因富集于胰岛peaks。此外,本研究共获得60,517个转录本,分为未表达(38,428个)、低表达(7,363)、中表达(7,363)和高表达(7,363)转录本。ATAC-seq 分析中的peaks大部分被注释到高表达的转录本上。
4. T2D和非糖尿病供体的胰岛开放染色质区域差异分析
有1078个peaks在T2D和非糖尿病胰岛供体之间存在差异,其在基因组不同区域的分布比例见图4a,其中有1044个peaks在T2D供体中富集。一些关键TF特异性motif显著富集在peak中(图4b)。

Bysani, M., Agren, R., Davegrdh, C., et al., ATAC-seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes. Sci Rep 9, 7785 (2019). doi:10.1038/s41598-019-44076-8
细胞分化由转录因子(TF)活性变化和转录的改变所驱动。目前,可通过探测染色质可及性去推断细胞类型间TF结合的差异研究这一过程。本研究采用转座酶可及染色质测序(ATAC-seq)分析,以描述拟南芥中茎尖分生组织(SAM)干细胞和分化的叶肉细胞在染色质可及性和TF调节网络方面的差异。
经实验室培养、转化的拟南芥植株
按一定条件培养植物;构建2种质粒(分别为CLV3和RBC基因与NTF基因的结合体);将质粒整合到植物中表达;拍摄记录转化后的植物顶端分生组织或叶片以确定NTF蛋白的表达和定位;用INTACT法分离细胞核;进行ATAC测序;数据分析与可视化。
1. 主要不同细胞的染色质开放区域注释
干细胞和叶肉细胞可富集到22,961个相同的THSs,单独在两类细胞表达的转座敏感位点THSs数量分别为5,283和2215个(图1b)。干细胞和叶肉细胞THSs在基因组不同区域上的分布类似(图1c)。此外,两类细胞的表达谱显示,干细胞的ATAC-seq信号在THSs中心区域最强,并向两侧急剧下降。且在叶肉细胞THSs表达较少的区域也能呈现较高的开放性,间接表明在叶肉细胞的染色质可及区域在干细胞也可获得,反之则不然。
2. 细胞富集型THS相关基因分析
结果显示染色质开放性更高的细胞类型可以富集到更多的THSs,位于这些区域附近的基因更有可能呈现出高表达。而与这些基因相关的ATAC-seq peaks大部分位于转录起始位点上游和转录终止位点下游(图2)。
3. motif分析与转录调控网络
本研究分别在干细胞和叶肉细胞富集到23个和129个差异表达转录因子(图3a)。结合干细胞IDD/GATA调控和在叶肉细胞高表达的4个TFs,确定其靶基因并绘制调控网络图(图3b),该图描述了TF间复杂的相互作用(图4)。
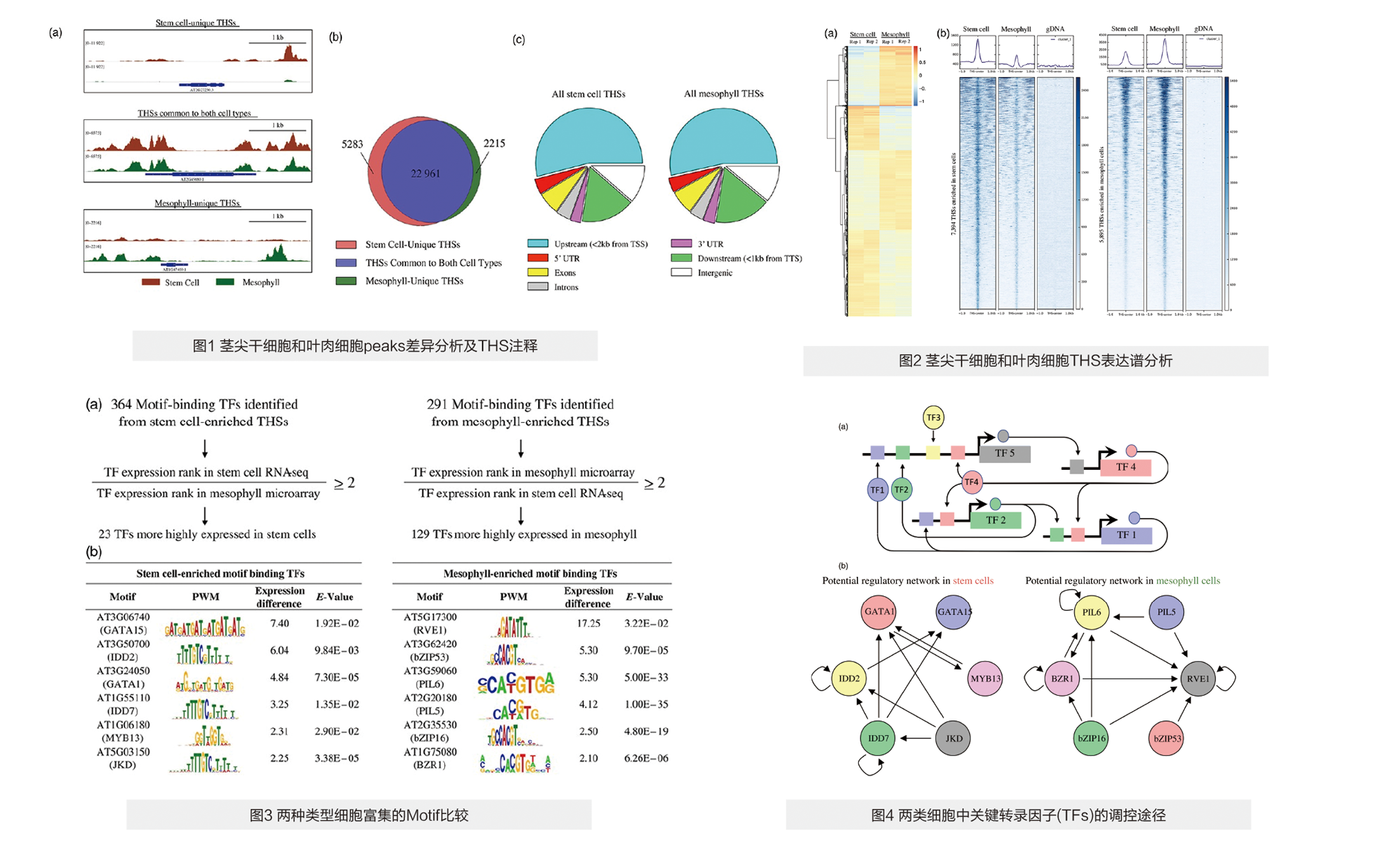
Sijacic P, Bajic M, McKinney EC, Meagher RB, Deal RB. Changes in chromatin accessibility between Arabidopsis stem cells and mesophyll cells illuminate cell type-specific transcription factor networks. Plant J. 2018;94(2):215-231. doi:10.1111/tpj.13882

ChIP-seq 是获取已知蛋白(转录因子)结合位点的方法,ATAC-seq主要是基于开放区域的motif基因序列来预测可能结合的转录因子。ATAC-Seq对样本起始量需求更少(500-50000),获取的信息更多,不但可做全基因组活性图谱,还可以做核小体定位和转录因子结合分析。
ATAC-Seq对细胞活性要求较高,送样前最好检测下细胞活性,尽量送活性(大于70%)较高的新鲜细胞,组织样本需先处理成细胞悬液, 组织样本也可承接,但需要提前评估。 风险样本类型:a) 结缔组织等骨细胞和纤维含量高的动物样本;b)根茎等纤维含量高、种子果实等糖含量高的植物组织样本,这些物质会降低细胞核提取效率;以及冻存时间超过2个月的样本。
建议每个分组中设置至少3个生物学重复,来获得更准确的结果,如生物重复间个体差异大的,可增加生物学重复数量。取样过程中,应减少人为造成的样品间差异,具体可以采取如下措施:取样时间、部位、处理条件、性别、年龄、操作过程等方面尽可能保持一致,否则会影响实验结果的可重复性和可信度。