宏基因组研究以环境中所有微生物基因组为研究对象,通过对环境样品中的全基因组DNA进行高通量测序,获得单个样品的饱和数据量,进行微生物群落结构多样性,微生物群体基因组成及功能,特定环境相关的代谢通路等分析,从而进一步发掘和研究具有应用价值的基因及环境中微生物群落内部、微生物与环境间的相互关系。构建的环境微生物基因集,可为环境中微生物的研究、开发和利用提供基因资源库。


农用杀菌剂增加了温室土壤抗生素抗性基因的丰度
期刊:Environmental Pollution 影响因子:5.714 发表时间:2020年4月
温室栽培长期施用杀菌剂导致土壤残留污染,改变土壤微生物群落。目前还不清楚残留杀菌剂是否影响土壤中抗生素抗性基因(ARGs)的多样性和丰度。可移动基因元件(MGEs)被认为是导致抗性基因在细菌间水平转移的重要原因。本研究利用宏基因组测序技术分析温室和山地土壤中杀菌剂的耗散及其对ARGs丰度影响,证实一些杀菌剂可能会通过MGEs介导的水平基因转移而提高温室土壤中ARGs的丰度。
温室土壤和山地土壤的微宇宙系统
室内构建温室和山地土壤实验环境;分别向土壤中添加杀菌剂(嘧菌酯、多菌灵等)和抗生素(金霉素),以无添加土壤为对照;在0、1、3、7、15、30和60天采集土壤样本;检测土壤样本中的杀菌剂和抗生素浓度;提取土壤DNA,宏基因组测序;数据分析与结果可视化。
1.温室土壤比山地土壤中含有更丰富多样的ARGs
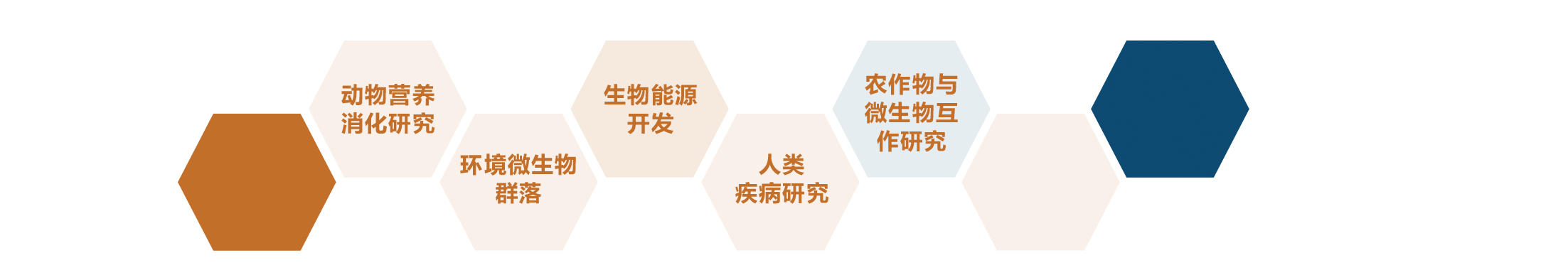
共检测出18类ARGs,其中温室土壤检出17类,山地土壤检出13类,温室土壤中ARGs的总丰度显著高于山地土壤。温室土壤中检测出ARGs 共计35-53种,山地土壤为10-21种,温室土壤中ARGs的多样性显著高于山地土壤(图1)。ARGs在两类土壤中的共现情况见图2。
2.多菌灵、嘧菌酯和百菌清的施用可以提高温室土壤中总ARGs的丰度
经过60天的实验室模拟,无论是在温室还是山地土壤中,添加三唑酮没有明显改变ARGs的丰度,但是多菌灵、嘧菌酯和百菌清的使用显著提高了土壤中ARGs的丰度。
3. 温室土壤中ARGs丰度与MGEs(如inti1、R485)的相关关系显著
本研究温室土壤中质粒和整合子的多样性及丰度均明显高于山地土壤,其中intI1在温室土壤中丰度很高,而在山地土壤中却很少。此外,温室土壤中MGEs与多种优势的ARGs存在显著的正相关,而山地土壤中与MGEs相关的ARGs数目非常少(图3)。
4. 进一步验证了ARGs和MGEs,如sul2和R485, sul1和转座酶的共现模式
本研究进一步通过宏基因组组装查找ARGs与MGEs的共存依据,在温室土壤中,共发现25对ARG与MGE位于同一个contigs中的现象,例如sul1与转座酶(图4)。这证明温室土壤中杀菌剂残留所带来的选择性压力可能会导致ARGs通过MGEs介导的HGT进行传播和增殖。
5. 温室土壤中ARGs主要由肠杆菌携带,山地土壤中则以放线菌为主
温室土壤中,共鉴定到26个潜在的ARGs宿主细菌属,其中Acinetobacter检出频率最高。在山地土壤中,共鉴定到21个ARGs的潜在宿主,其中最主要的宿主是Nocardia和Streptomyces,与温室土壤一样,vanR基因同样是山地土壤中宿主范围最广的ARG(图5)。
Zhang HP , Chen SY , Zhang QK , Long ZN , Yu YL, Fang H. Fungicides enhanced the abundance of antibiotic resistance genes in greenhouse soil. Environmental Pollution. 2020. doi.10.1016/j.envpol.2019.113877
宏基因组测序鉴定HPV16感染者阴道微生物组的变化
期刊:Frontiers in Cellular and Infection Microbiolog 影响因子:4.12 发表时间:2020年6月
微生物群失衡与癌症发展的关系是目前研究的热点之一。长期感染人乳头瘤病毒(HPV)会引发宫颈癌,但目前对HPV感染中的微生物组组成和功能知之甚少。本实验使用宏基因组测序技术鉴定了HPV16阳性组和HPV阴性组的阴道样本的成分和功能变化,发现HPV16阳性组阴道微生物群的组成和功能发生了改变,表明阴道生态失调可能与女性生殖道HPV感染有关。
在2017.2-2018.11期间使用HPV检测和细胞学进行宫颈癌筛查后进行阴道镜检查的2251名未怀孕育龄妇女
筛选符合要求的女性作为实验对象;从阴道穹窿和宫颈附近取4个无菌拭子样本进行实验分析;样本分为HPV16阳性组和对照组(HPV阴性和细胞学正常的女性);分别对样本中的微生物进行DNA提取与测序;数据分析与结果可视化;用定量PCR检测微生物标记并作出评价。详细实验流程见图1。
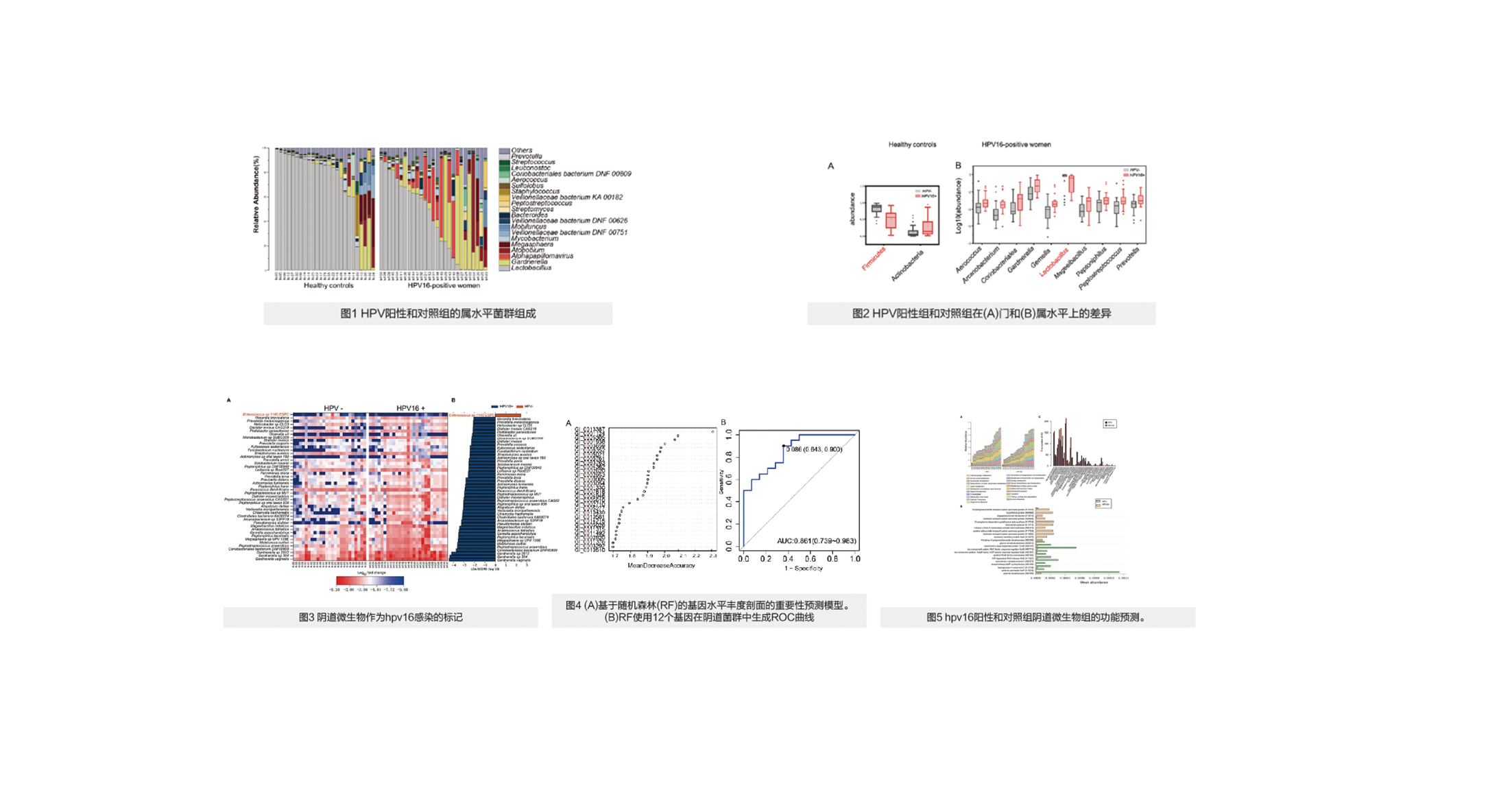
1.HPV16阳性妇女阴道菌群的分类变化
尽管HPV16阳性组与对照组均以乳酸杆菌属、加德纳菌属占绝对数量优势(图1)。但进一步研究发现HPV16阳性组厚壁菌门(图2A)、及属于厚壁菌门的乳酸杆菌属丰度显著降低(图2B)。相反,阳性组放线菌门以及属于放线菌门的加德纳菌属的丰度显著增加(图2B)。
2. HPV16感染生物标记物分析
共44种生物标记物在HPV16阳性组样本中富集,仅1种生物标记物在对照组富集(图3)。本研究使用了三种类型的生物标记(12个基因,17个属和7个物种)构建随机森林集成学习法区分HPV16阳性组和对照组,结果显示该方法对HPV16阳性具有较高的预测能力(图4)。
3.与HPV16感染相关的阴道微生物基因
通过Metastats分析,确定了参与88个通路的378个KEGG同源序列 (KOs),在HPV16阳性组和对照组之间差异显著。将在同一途径上序列和功能高度相似的蛋白进行分组,共鉴定出 22 个 KOs,且在HPV16阳性和对照组阴道菌群中的丰度差异显著(图5B)。进一步分析发现,HPV16 阳性组在代谢和膜运输中富集,而在多糖生物合成和代谢、复制和修复中耗尽(图5C)。
4. 评价HPV16感染的生物标记物
随机选择两个16阳性的富集基因标记进行定量PCR检测,结果与宏基因组测序结果具有很强的相关性,证实了方法的可靠性。
Yang Q, Wang YP, Wei XY, Zhu JW, Wang XY, Xie X, Lu WG. The Alterations of Vaginal Microbiome in HPV16 Infection as Identified by Shotgun Metagenomic Sequencing. Frontiers.. 2020. doi: 10.3389/fcimb.2020.00286

不可以,如果宿主的基因组序列在环境DNA中的量比较多,测序之后,我们没有办法通过已知的宿主基因组的序列来去污染,会对最后的分析结果造成很大影响,而且可用的数据量会很少;但是如果在提取的过程,宿主基因组的污染的量很少,后期的数据分析还是可用的,但是会存在一定的风险。
宏基因组样本量需求跟研究目的直接相关。如果侧重于功能挖掘,一个或几个样本即可;如果侧重于组间差异分析,如疾病相关的肠道菌群研究,一般需要较大的样本量,一般每组样本在50个左右,可参考对应的经典研究案例。