Small RNA(包括 miRNAs、siRNAs 和 piRNAs 等)是生命活动重要的调控因子,在基因表达调控、生物个体发育、代谢紊乱及疾病发生和发展等生理过程中发挥重要作用。miRNA 是一类内源性的、长度约 18-25 nt、具有调控功能的非编码RNA,通过和靶基因碱基配对引导沉默复合体降解 mRNA 或阻碍其翻译。Small RNA 测序技术不受物种限制,既能鉴定特定条件下表达的 miRNA,又能预测发现新的 miRNA,对于疾病的发生发展研究有重要意义。



微小RNA(miRNA)是一类长度19-25个核苷酸的内源性非编码调控RNA,其通过降解mRNAs或抑制mRNAs翻译来参与细胞生物学功能调节,包括细胞发育和分化、代谢、细胞周期和凋亡等及重要炎症调节。急性淋巴细胞白血病(acute lymphoblastic leuke-mia, ALL)是一种起源于淋巴细胞的B系或T系细胞在骨髓内异常增生的恶性肿瘤性疾病。miRNAs异常表达对于ALL的发病机制与临床应用有至关重要的作用,miRNAs已迅速成为 T-ALL潜在的治疗靶点。然而,目前对T-ALL的miRNA转录研究较少。
骨髓样本来自34例儿科T-ALL患者和5位年龄小于18岁的健康骨髓供体。使用密度梯度离心方法分离骨髓样本的单核细胞,后进行免疫磁分离,以获得T-ALL细胞和正常成熟的T淋巴细胞(对照)用于RNA分离。此外,本研究使用GSE89978数据进行联合分析。
对34例实验组和5例对照组进行miRNA测序并分析,后联合公共数据分析,其分析流程参照miRge。其差异miRNA结果由RT-qPCR验证,通过验证的miRNAs分析其靶基因和参与的KEGG/GO通络。
1. miRNA表达谱分析
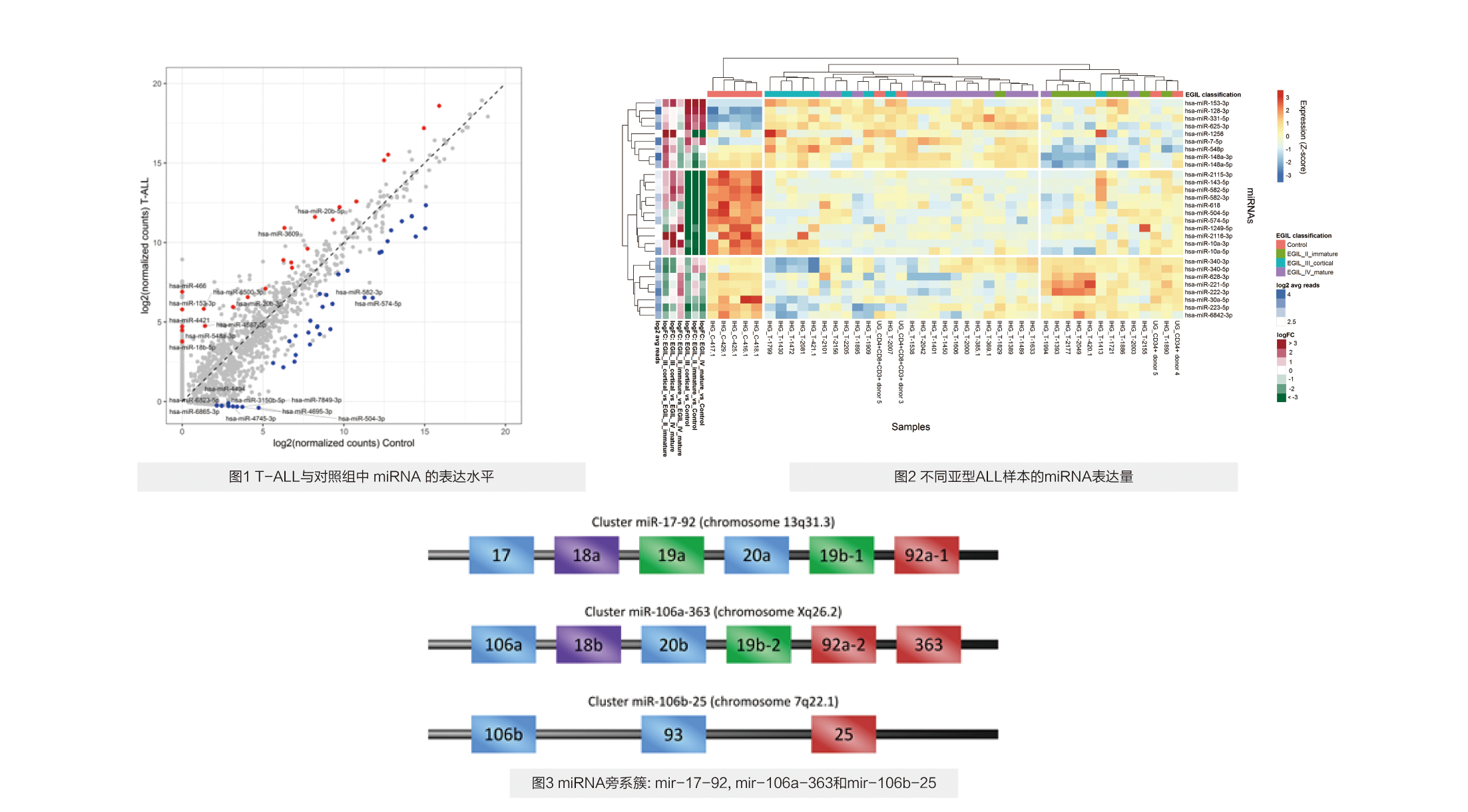
对34例T-ALL组与5例对照组分析结果显示有1462个miRNAs表达,其中452个仅在T-ALL实验组中表达(图1)。差异分析共鉴定出61个差异表达miRNAs,其中hsa-miR-128-3p,hsa-miR-181a和hsa-miR-181b已在其它致癌基因研究中报道。联合分析中,实验vs对照组中T淋巴细胞和胸腺细胞中的miRNAs分析显示有103个差异表达miRNAs。在T-ALL或正常成熟T淋巴细胞和胸腺细胞中差异过表达的miRNA,分别代表候选致癌或抑癌miRNA。此外,对34例T-ALL组/5例对照组的miRNA分析结果展示出不同亚型T-ALL呈现不同的miRNA表达谱(图2)。
2. 靶基因预测和通路富集分析
对在T-ALL与对照组差异表达的61个miRNA进行靶向预测,通过筛选,最后定义了225个预测靶标。这个靶向基因在KEGG、GO通路富 集在与T-ALL发病机理相关的生物学通路中,包括白介素6–介导的信号传导,mTOR信号传导和凋亡调控。研究最终专注于hsa-mir-106a-363簇,并在功能上验证了hsa-miR-20b-5p和hsa-miR-363-3p与它们的预测靶标(PTEN,SOS1,LATS2)的3'端非翻译区的直接相互作用,深度参与调节细胞凋亡(图3)。 hsa-mir-106a-363是直接致癌hsa-mir-17-92簇的旁系同源物,在T-ALL的发病机理中尚未确定。本研究为miRNA-mRNA相互作用在T-ALL中的功能分析提供了坚实的基础和数据资源。
Dawidowska M, Jaksik R, Drobna M, et al. Comprehensive Investigation of miRNome Identifies Novel Candidate miRNA-mRNA Interactions Implicat- ed in T-Cell Acute Lymphoblastic Leukemia. Neoplasia. 2019;21(3):294-310. doi:10.1016/j.neo.2019.01.004
鉴定辣椒疫霉感染诱导拟南芥miRNAs的变化
期刊:Frontiers in Microbiology 影响因子:4.235 发表时间:2020年6月
Phytophthora capsici是一种土传致病性卵菌,可引起50多种植物的严重枯萎病和果实腐烂,包括辣椒、番茄、黄瓜和其他重要的商业作物。最近的研究表明,辣椒-拟南芥系统是分析卵菌-植物相互作用的模式病理系统。探索宿主-病原菌之间的相互作用是提高我们对致病性的分子基础的理解和制定疾病管理策略以保护食品生产免受辣椒疫霉感染的第一步。内源性sRNAs是植物免疫系统对各种病原体作出反应的一种普遍的调节机制。然而,辣椒疫霉侵染对拟南芥内源性sRNAs的影响尚未见报道。
将哥伦比亚生态型拟南芥(Col-0)置于22℃土壤,长日照环境下生长。Col-0对辣椒疫霉LT263高度敏感。用10 μL 椒悬浮液(实验组)或MgCl2(对照组)处理3周龄Col-0植株的叶片背面。在0(对照组)、3、6、12和24 小时收集叶片,液氮冷冻提取RNA。
本研究对实验组和对照组进行sRNA测序。为了验证sRNA-seq结果的可靠性,研究使用northern印迹分析和qRT-PCR研究了在4个感染阶段上调的5个sRNA的表达及其潜在的靶基因。
1. miRNA序列分析
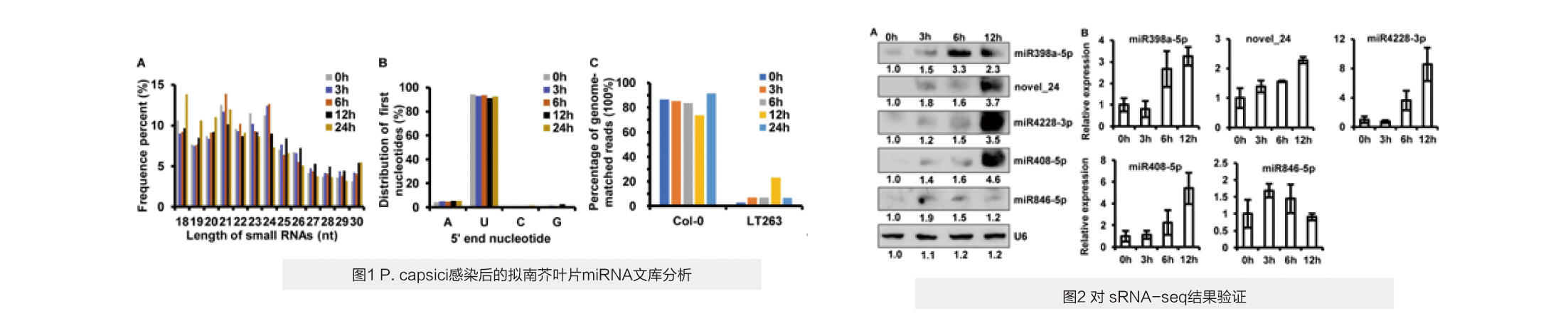
对miRNA序列的分析表明,长度为21/24 nt的sRNAs在四个感染阶段含量均最高(图1A)。长度为21/24 nt miRNA序列的第一个核苷酸中90%以上是尿嘧啶(图1B),表明植物sRNAs的分布高度一致。在0、3、6、12和24个hpi文库中,分别有86.73、85.15、83.60、73.63和91.55% reads比对到Col-0基因组上(图1C)。此外,鉴定出293个已知miRNAs和6个潜在的新sRNAs(miRNAs或siRNAs),其中33个miRNAs在4个不同感染阶段有差异。
2. miRNA表达谱分析
与对照组相比,分别有23、28、29和30个miRNAs在3、6、12和24 hpi分别上调。此外,有27、23、30和26个miRNAs在这四个感染阶段下调。其中,有19个和14个miRNAs在所有四个感染阶段分别上调和下调,表明约30.9%的新鉴定miRNAs在辣椒疫霉感染期间持续表达。
3. 差异miRNA和其靶基因验证
sRNA-northern杂交结果与sRNA-seq数据基本一致。在辣椒疫霉菌感染后,所有5个上调miRNAs的表达量显著增加到最高水平(图2A)。但与sRNA-seq分析结果不同的是,northern印迹分析显示,在12hpi时,novel_24、miR4228-3p和miR408-5p的累积量大于其他时间点(图2A)。茎环qRT-PCR结果与sRNA-seq和northern杂交结果一致。与对照组相比,在12hpi时,novel_24、miR4228-3p和miR408-5p的转录丰度最高,而miRNA846-5p的转录丰度在3hpi时最高(图2B)。
4. 基因富集分析
GO和KEGG富集分析表明,差异表达miRNAs的潜在靶基因都在淀粉和糖代谢、剪接体和植物-病原菌相互作用等途径中富集,说明剪接机制和致病相关蛋白在辣椒疫霉菌感染反应中起重要作用。
ZHU, Xiaoguo, et al. High-throughput sequencing-based identification of Arabidopsis miRNAs induced by Phytophthora capsici infection. Frontiers in microbiology, 2020, 11: 1094. doi: 10.3389/fmicb.2020.01094

可以。目前可抽提细胞、血浆、血清样品的外泌体miRNA。
样品类型包括总RNA,组织细胞样品,血清血浆样品以及切胶回收的Small RNA样品等。其中总RNA提取不要使用过柱试试剂盒。
种子区是成熟 miRNA 5’端第 2-8 位序列,是 mRNA 结合中最关键的序列。
miRNA 数据库包括miRBase、miRDB、PMRD等。目前 miRBase (http://www.mirbase.org/)已升级至 22.0 版本,囊括了 223 个物种的miRNA。 PMRD (http://bioinformatics.cau.edu.cn/PMRD/)是一个专门针对植物 miRNA 的数据库常用miRNA靶基因预测网站或软件有RNAhybrid、miRanda、microRNAorg、TargetScan、 Pictar 等。