全基因组重测序(WGS)是对已知基因组序列的物种进行不同个体的基因组测序,并在此基础上对个体或群体进行差异性分析。它可以获取最全的基因组信息,找到大量的单核苷酸多态性位点(SNP),插入缺失位点(InDel)、杂合性缺失(LOH)、拷贝数变异(CNV)以及基因组重排导致的结构变异位点(SV)。配合比较基因组学分析、群体遗传学分析、进化分析和计算生物学分析方法既可以用于深入探索疾病基因组的奥秘,也可以高效识别动植物的性状连锁区域,为遗传育种提供科学依据和技术支撑。


基因组测序分析确定EB病毒亚型与高发病鼻咽癌的关系
期刊:Nature Genetics 影响因子:27.603 发表时间:2019年6月
鼻咽癌(NPC)是一种具有独特地理分布的恶性肿瘤,在全球大多数地方很少见,但在中国华南、东南亚、地中海和格陵兰岛等局部区域高发。其中,广东地区鼻咽癌的发病率位居首位,每年10万人中有20-40人被诊断为鼻咽癌,是其它低发病率地区近20倍。EBV是一种与NPC、胃癌、淋巴瘤有关的人类病毒,对自发性淋巴母细胞系和EBV相关疾病的EBV基因组测序表明,不同地理起源的EBV分离株之间存在重要的基因组变异。因此,研究者希望通过EBV全基因组测序(WGS)寻找我国南方NPC高风险EBV亚型,为确定NPC高危人群提供依据。
来自诊断为EBV相关癌症患者体内分离的215株EBV,来自中国NPC流行区和非流行区健康对照体内分离的54株EBV
利用全基因组测序(WGS)分析269例来自鼻咽癌高发区和低发区的鼻咽癌患者及健康对照中的EB病毒全基因组信息,对比鼻咽癌和健康人来源的EB病毒全基因组信息。
1.EBV全基因组测序和 Fine-mapping确定与NPC相关的EBV突变
研究者对269株EBV分离株进行全基因组测序,PCA分析结果表明,EBV沿PC1的分布是连续的(从非洲、欧洲到亚洲),沿PC2的分布表现出部分分离的模式(亚洲NPC流行区、NPC非流行区)。从基因型看,BALF2的突变SNP162215、SNP162476、SNP163364为高风险基因型。
2. 探索三种SNP与NPC的关系
对156例SNP和47例对照进行全基因组关联分析,发现SNPs 162507C>T, 162852G>T 和 162215C>A 在NPC患者全基因组中呈显著差异。进一步验证三个SNP与NPC的相关性,发现与162476T> C和163364C> T相比,SNP162215_C不太可能是与NPC相关的突变位点。

Xu Miao,Yao Youyuan,Chen Hui et al. Genome sequencing analysis identifies Epstein-Barr virus subtypes associated with high risk of nasopharyngeal carcinoma.[J] .Nat. Genet., 2019, 51: 1131-1136
阿拉伯联合酋长国混合人口的四个代表的全基因组测序
期刊:Frontiers in Gentics 影响因子:3.258 发表时间:2020年7月
阿拉伯地区有着独特的人口结构和种族多样性,但是关于人口层面相关的深入遗传研究很少。使用全基因组关联分析来研究人群的遗传结构,可提供有关基因组原型的深入知识。本研究是1000个阿拉伯基因组计划的一部分,对四名阿联酋国民的全基因组进行基因组变异分析。这四名阿联酋国民代表不同的亚人口存在,并构成历史种族混合阿联酋人口。
来自四名阿联酋国民的唾液样本
利用全基因组测序(WGS)分析4名受试者的唾液样本,进行全基因组测序读数比对,SNP和Indels注释,SV识别,估计4个WGS单倍群的父系和母系的祖先,并估算了4个样品的遗传祖先,此外对4个样本的基因组之间的距离以及来自HGDP世界人口的基因组之间的距离进行了系统发育定位,变种的验证,从基因分型数据对本地等位基因频率进行注释。
1.四个阿联酋样本概述
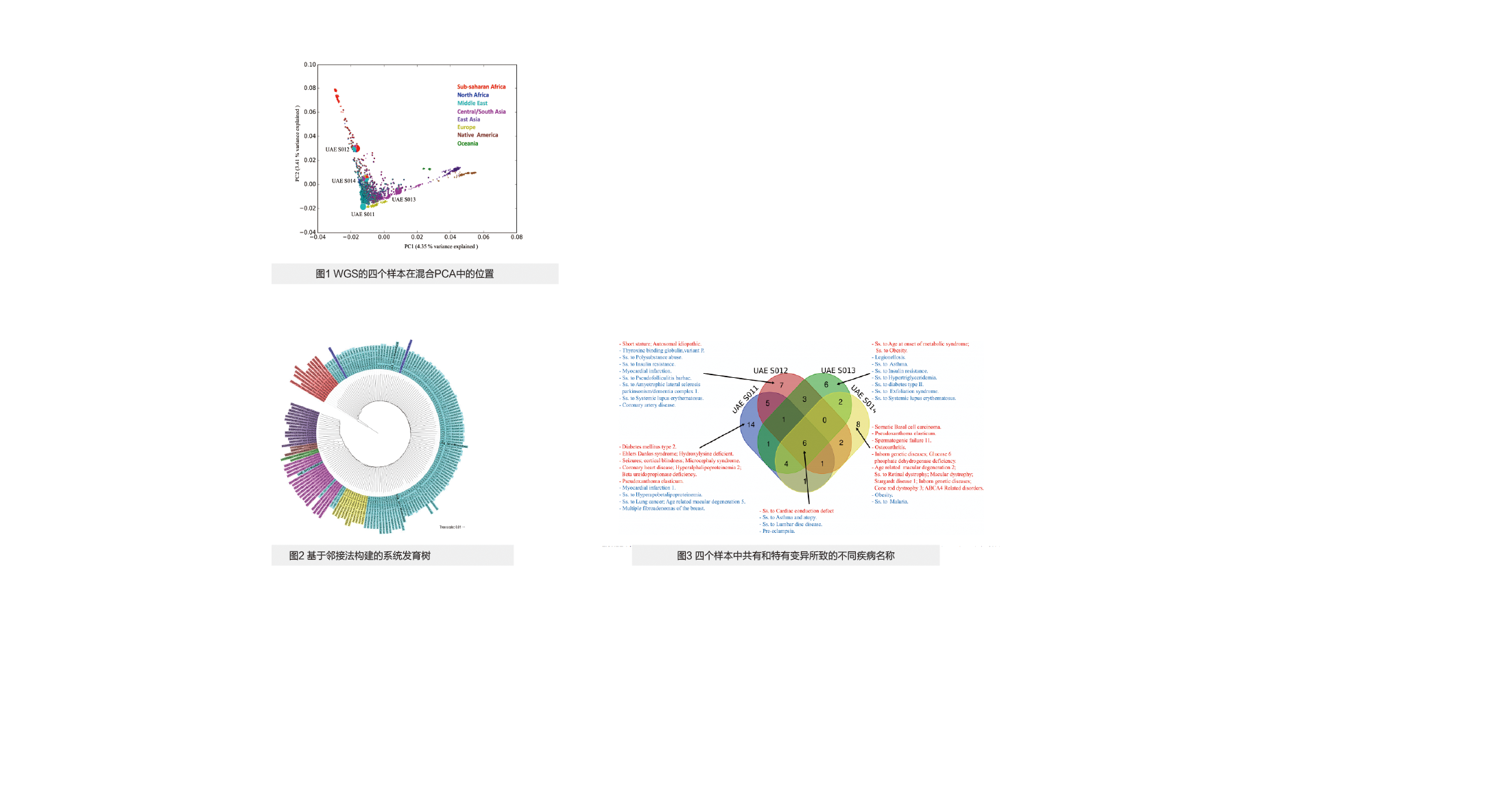
根据混合程度和亚群中心性,选择四个WGS中阿联酋不同亚群代表的样本。从PCA图中可以看出这四个样本在HGDP(人类基因多样性工程)中的情况(图1)。
2.四个样本的变异分析
这四个样本平均每个样本具有4,427,749个确定的变异体。在这些变异中,平均80%为SNP,20%为InDel。杂合子和纯合子变异的比例分别为65.4-57.4%和42.6-34.6%。进一步确定三个男性个体分别属于Y-单倍型群:J1(UAE S011),E1(UAE S012)和J2(UAE S014)。此外,UAE S011和UAE S014拥有相同的H2mtDNA。
3. 进化树分析及SV分析
使用包含四个主体的邻接方法重建系统发育树(图2),发现UAE S011和UAE S014样本属于中东的阿拉伯人群组,UAE S012也归入中东群组,但与撒哈拉以南非洲集团更接近,而UAE S013处于中/南亚群组的中心位置。此外,SV总数为15677至20339,只有约13.5%是新发现的。
4. 与特定疾病相关的变异
通过将具有潜在易感性的变异与疾病列表关联分析,本研究描绘出个人基因组的基因型与疾病之间的关联。图3显示了四个样本中共有或特有的,且被认为是可致病(红色)或被评估为危险因素(蓝色)的变异,例如,共有变异可致的疾病包括心脏传导缺陷等。
Daw Elbait G, Henschel A, Tay GK, Al Safar HS. Whole Genome Sequencing of Four Representatives From the Admixed Population of the United Arab Emirates. Front Genet. 2020;11:681

每个样本推荐的数据量与样本类型和要做的信息分析内容相关。例如关注个体样本的SNP,对SNP的准确度和覆盖度要求比较高,一般推荐测序深度>30X,对于稀有变异测序深度还要进一步提高;用于研究群体结构的样本,测序深度推荐10X以上;纯合样本混样检测等位基因频率,推荐平均每个样本的测序深度在1X以上,混合样本测序深度不低于30X;DH和RIL群体构建Bin Map,子代群体测序深度可以测序1X/样本。
样本量大小与样本类型和研究目的相关。例如进行群体进化研究推荐30个样本以上,因为从统计学上说30个以上才属于大样本;对于进行基因挖掘的项目来说,无论是利用自然群体进行GWAS分析或是用家系群体进行连锁分析,都是群体越大越好,一般的情况下进行GWAS分析的样本推荐300个样本以上,对于家系群体推荐200个以上。